- EJPPS

- Jul 5, 2022
- 4 min read
Updated: Jul 14, 2022
Opinion Article | Open Access | Published 14th July 2022
RFC and Alternative Validation (ALT VAL)
Author: Kevin Williams, bioMerieux
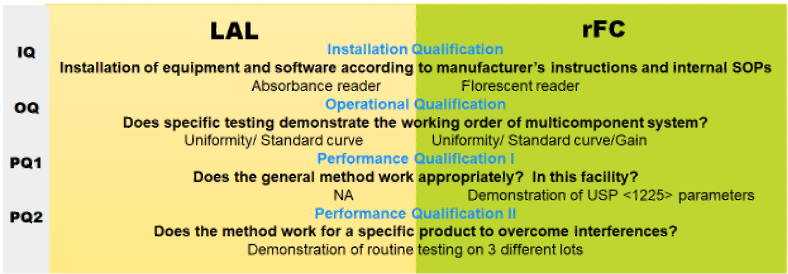
I. Overview
Most of the activities required for alternative validation (ALT VAL) are activities associated with setting up any new test in a laboratory and include (i) installation qualification (IQ), (ii) operational qualification (OQ), and (iii) performance qualification (PQ). ALT VAL as required by USP <1225> and when presented with methods that are as analogous as LAL and recombinant Factor C (rFC) it is much "simpler than you think".
This paper presents an overview in an extremely condensed form. The complete protocol to be performed as well as the supplier performed validation data are both available from bioMérieux.
II. Definitions
The definitions governing each stage of qualification are given below:
IQ governs the performance and documentation of the installation of the reader and software as a multicomponent system. It ensures that the respective manufacturer provided instructions are followed by users that are also following existing procedures governing such activities. If a user does not have a fluorescent reader SOP then a draft must be put together. bioMérieux supplies generic copies for users to specifically elaborate.
OQ, once the IQ is complete (performed, documented, approved) then the user must demonstrate and document that the installation has been successful on the operational level. Typically this will include testing that will be used subsequently in the periodic maintenance (PM) activities going forward. These tests (eg. uniformity test etc.) come with stated acceptance criteria given in newly drafted specific SOPs that the user generates (from supplied generic documents).
GAIN test -For fluorescence, a “gain” test is needed. A gain test simply runs a 0.5 EU/mL standard set to calibrate the dRFU (delta relative fluorescent units) onto the best part of the range of the photomultiplier. This is performed for each reader and RFC lot and used until the lot or reader is changed.
PQ demonstrates the holistic performance of real tests using (mock) samples. Some find it useful to think of PQ as existing in two parts designated as: Part 1 (PQ1) which is the method validation as this is a new method compared to LAL. This is where accuracy, precision, ruggedness, specificity etc. are demonstrated according to a detailed protocol (supplied by bioMerieux).
III. Rationale
The FDA (2012 Q&A Guideline) recommends documenting the “rationale” for switching from LAL to rFC. There are no shortages of valid rationales to choose from including:
Supply chain sustainability – also viewed as “Global availability”. One’s affiliates should not suffer because they are further away from Delaware Bay.
Environmental conservation (Going “Green”). There is no need to apologize for wanting the animals of the planet to survive the onslaught of man, including horseshoe crabs and the shorebirds that depend upon their eggs to support their migratory patterns.
Elimination of potential false positive values from beta-glucans and/or cellulosic residues (from pharmaceutical manufacturing filters)
Increased throughput via automation or semi-automation. LAL automation cannot match the 120 samples per hour that can be achieved using the fluorescent end-point method.
Analyst reduction – a test using ENDOZYME II GO, where standards and positive product controls are already added into the plate (dried in) cuts the time and effort needed to perform an endotoxin test significantly.
Reduction of repeat testing – not preparing a vial of CSE (GO Plate) and an expectation of a standard curve that works every time will reduce repeat testing.
For BIOLOGICS, a simplification of the test reagent milieu may help investigations in method development for a specific molecule interaction given that rFC contains a single protein (rFC) versus a myriad of various proteins from the harvested HSC hemolymph (Factor G, Factor B, Proclotting enzyme, coagulogen, serpins, antimicrobial peptides, etc.).
Cost savings, since LAL is supplied in discrete units (vials of 2.6 or 5.2 mL when reconstituted), the optimum amount to be used often brings waste. RFC on the other hand, as a liquid reagent, can be used in the exact proportion to the number of samples to be tested.
IV. Getting Started
The simplest way to begin testing is to apply the test to purified water, raw materials and container/closure testing. For finished drug testing, the pre-submission of alternative validation can be more time consuming and often requires submission to multiple regulatory bodies. In starting with non-drug testing, no pre-submission or “permission gaining” exercise is required when using the new test. Just perform the validation and document it for inspection-ready access if or when FDA inspectors ask for it. Note that rFC is compendial in Europe (Chapter 2.6.32). In the US one can also reference the MAPP procedure by which other pharmacopeias can be referenced for FDA submission (1). By switching this testing prior to product testing it covers 85-90% of all testing and thus helps the user support his/her supply chain and sustainability goals (rationale) without delay. Also, a recent user has demonstrated “non-inferiority” testing by comparing LAL/rFC and present a good case for including this method in a validation in terms of reasonable acceptance criteria (2).
Manual of policies and procedures, Center for Drug Evaluation and Research. Acceptability of standards from alternative compendia (BP/EP/JP). MAPP 5310.7 Rev. 1.
Validation strategy for new recombinant factor C users, Dre et al., Amer. Pharm. Rev., Feb. 2022.


Comments