- tamsinmarshall4
- Jul 11, 2025
- 15 min read
Peer Review Article | Open Access | Published 11th July 2025
Current Regulations For Variation Filings For Registered Products In Europe And Frequently Submitted Variations- Part 1
Arunodaya B S,1 Gaganashree T V*, Balamuralidhara V2, Chinmayee U Gowda3, JSS College of Pharmacy, JSS Academy of Higher Education Research | EJPPS | 302 (2025)| https://doi.org/10.37521/ejpps30205 |
Abstract
The requirement to maintain current and updated dossiers is a need across the pharmaceutical industry, regardless of a product's route or country of registration. Marketing authorization holders have a major duty for post-approval lifecycle management operations, regardless of whether changes are brought on by advancements in technology and science or decreased costs (MAHs). The procedures for filing and processing variants are starting to converge as regulatory bodies throughout the world modernise. The fundamental principles of regulatory management of product lifecycles are understanding the requirement for variations and preventing pointless variations. This supplement for continuing education is the first of a series on lifetime learning that will appear periodically. examines the most prevalent variants. It compares the European process for Type IA, Type IB, and Type II variants to the US approach, outlining important parallels and differences in line extensions, grouping, and task sharing methods. A directive gives forth a standardized list of expected variants with categorization codes in addition to the European legislation that specifies variation kinds. Since the Mutual Recognition Procedure (MRP) was put into place in 1998, there is a set list of modifications for European MAs. However, several EU member states at the time did not completely ratify the legislation controlling European variant processes at the national level. Legislation has periodically been updated and in the most recent update, in August 2013, implementation was made mandatory at the national level and the variation process has been completely harmonized across the EU.
Keywords – Variation, Extension, Timeline, EMA, Procedures.
1. INTRODUCTION
Marketing authorization or registration of medicinal products is a dynamic process. It involves pre-marketing assessment of drug dossiers to verify quality, safety and efficacy based on the existing evidence and thereafter a change depending on emerging issues that arise during the lifetime of the product. The Marketing Authorization Holder is therefore required to take into account technical and scientific progress of a registered product throughout its lifetime. The holder is required to make any amendment that may be required to enable the registered product be manufactured and checked by means of generally accepted scientific methods.
Any changes to registered products (variations) may involve administrative and/or more substantial changes and are subject to approval by the relevant regulatory authority in the European Union. Procedures for the implementation of the different types of variations are established to facilitate the tasks of both Marketing Authorization Holders (MAHs) and regulatory authorities, and to ensure that variations to a medicinal product do not pose public health risks. The applicable guidelines outline the conditions to be fulfilled by applicants and the type of documentation required before a variation can be approved. The variations are categorized under defined types and schedules as per EU legislation, such as Commission Regulation (EC) No 1234/2008, which governs variations to the terms of marketing authorizations in the EU.
Schedule I: Lists minor changes. These are classified by the type of change as such and the conditions which frame this type of change. Whenever the conditions are not kept, the change may either become a major change or may even make a new application necessary.
Schedule II: Lists examples of major changes.
Schedule III: Lists types of changes which make a new application necessary.
Schedule IV: Lists stability requirements for variations and changes to registered finished pharmaceutical products (FPPs)
Commission Regulation (EC) No 1234/2008 of 24 November 2008 concerning the examination of variations to the terms of marketing authorizations for medicinal products for human use and veterinary medicinal products (1) (‘the Variations Regulation’) governs the procedure for the variation of marketing authorizations. It has been amended by Commission Regulation (EU) No 712/2012
UK Withdrawal from the EU and the Regulatory Implications (2025 Update)
The United Kingdom (UK) formally withdrew from the European Union (EU) on 31 January 2020, an event widely referred to as Brexit. This marked the end of the UK's participation in EU regulatory systems, including the European Medicines Agency (EMA). Following the withdrawal, there was an 11-month transition period during which EU law continued to apply in the UK while both sides negotiated future arrangements. This transition period ended on 31 December 2020, after which the UK began regulating medicinal products independently through its national agency, the Medicines and Healthcare products Regulatory Agency (MHRA).
A special case exists for Northern Ireland (NI), which shares a land border with the Republic of Ireland (an EU member). To avoid regulatory barriers and preserve the peace process outlined in the Good Friday Agreement, Northern Ireland remains aligned with specific EU rules on goods, including medicinal products. This arrangement was initially governed by the Northern Ireland Protocol and was later replaced by the Windsor Framework, agreed in February 2023. Under the Windsor Framework, Northern Ireland continues to follow EU pharmaceutical legislation for the marketing and movement of medicines, while the MHRA remains the responsible authority for granting and overseeing marketing authorizations in the region. This dual alignment ensures that medicines can be supplied freely within Northern Ireland and to both Great Britain and the EU Single Market.
As a result:
· Any marketing authorization variation filed for the European market continues to apply in Northern Ireland.
· At the same time, manufacturers must also comply with MHRA requirements for marketing in Great Britain (England, Scotland, and Wales).
For example, if a company wishes to implement a Type IB variation (e.g., changing a batch size beyond 10-fold), it would need to:
· Submit the variation under Commission Regulation (EC) No 1234/2008 for the EU and Northern Ireland, and
· Submit a separate variation application to the MHRA for Great Britain, which may follow different classification or data requirements.
This unique arrangement introduces added complexity for Marketing Authorization Holders (MAHs), who must now be aware of regulatory divergence between the EU, Northern Ireland, and Great Britain, especially in post-authorization changes and lifecycle management.
National Procedure (NP): If an applicant aims to procure a Marketing Authorization (MA) in a chosen EU member state, it is required to submit a Market Authorization Application (MAA) to the respective state’s competent authority through the NP.
Mutual Recognition Procedure (MRP): In MRP market authorization granted in one EU member state is recognized in other EU member states. MRP is applicable only when the manufacturer has already obtained market authorization in one of the EU countries. The regulations for market authorization through MRP are established in the Directive 2001/83/EC. If an application for MRP is submitted to more than one EU country, it must be ensured that all the applications are identical. The country evaluating the application is called as the Reference Member State and is responsible for notifying the other concerned Member States regarding the status of the application.
Decentralized Procedure (DCP): DP is applicable for medicines which have not yet been authorized in the EU. For these medicines, manufacturers can apply for simultaneous authorization in several EU member states. The procedure falls under the Directive 2004/27/EC. In the DP, anyone-member state can take the initiative of evaluating the application.
Centralized Procedure (CP): The CP allows manufacturers to submit a single Market Authorization Application (MAA) to the EMA. CP falls under the Regulation (EC) 726/2004.[2]
1.1 BACKGROUND TO CHANGES
Commission regulations (EC) 1084 and 1085/2003 – (CP, MRP products – not nationally approved) Commission – Review project launched in 2006 “Better Regulation”
Objectives
- Clear, simpler, more flexible
- Reduce administrative burden
- Adapt to ICH concepts (Q8, Q9, Q10)
- Further harmonies handling of variations in EU
2. EUROPEAN VARIATIONS SYSTEM-EVOLUTION
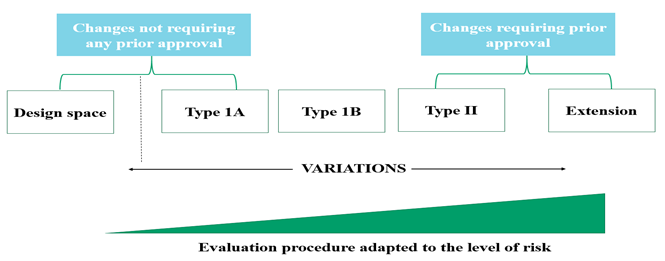
3.Types of Variations
Administrative
In their classification criteria, EU authorities even classify "administrative" as a category, although in other countries they fall into the lowest variation group and exhibit substantial crossover with small alterations, such as new addresses. The fact that these modifications can be executed without permission is acknowledged by several bodies. Administrative changes that require prior approval are time-consuming for agencies and expensive for business. Additionally, the modification might need to occur before submission in some circumstances, such as when a marketing authorization holder (MAH) changes their address.
Minor
Minor variations are often thought to either not affect the product's quality or have a very low likelihood of doing so, which lowers the risk. As a result, less agency review is necessary, which cuts down on the amount of time needed. Agencies have established methods to allow the smallest adjustments to be adopted before review as regulatory frameworks have evolved. To reduce the amount of assessment necessary, the EU, for instance, requires that when a Type IA variation is filed, the MAH specify which of a pre-defined set of requirements applies to its alteration. Many agencies have well-established documentation requirements for small alterations that must be completed before variations are filed. This means that MAHs are aware of what is anticipated prior to a submission and are able to gather enough supporting information. As a result, assessment periods are shortened since assessors are less likely to need to ask applicants for more information.
Major
When significant changes to product registration are necessary, they are carefully restricted, necessitating in-depth examination and review since they are anticipated to have an influence on a product's quality and efficacy. The MAH must show that the product will continue to be as effective and high-quality. Comparative data, which must credibly demonstrate that the proposed modifications do not lower product quality, is a crucial need for such improvements. Assessment durations for these variants are typically substantially longer since agencies thoroughly evaluate applications and frequently ask for further information and clarification on certain issues.[3]
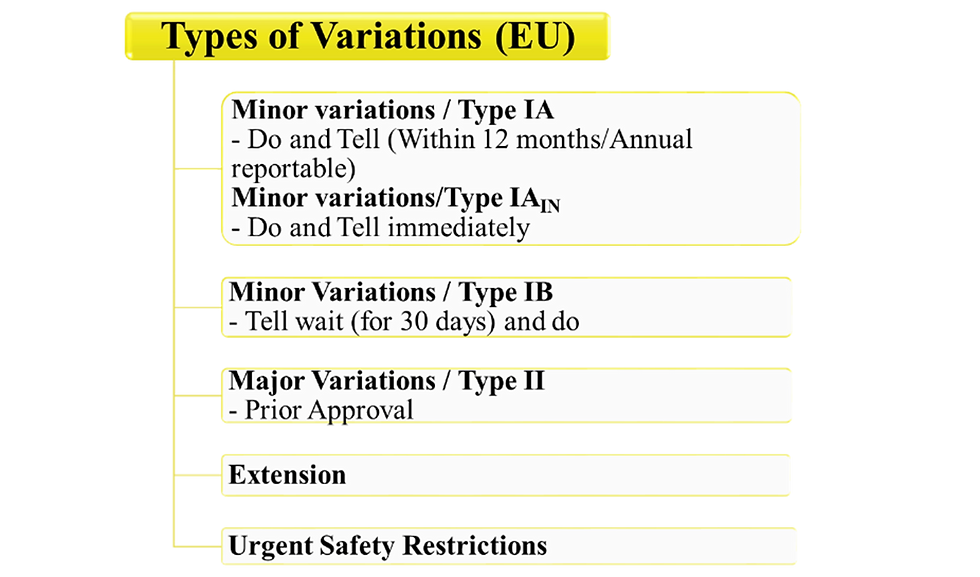
3.1 Minor Variation or Type IA:
Variation which has only minimal or no impact on the quality, safety or efficacy of the medicinal product. Do not require prior approval for implementation
These are again classified into 2 subcategories:
Type IAIN - Requiring immediate Notification
Type IA - Not requiring immediate notification. May be submitted within 12 months after implementation (Annual reportable)
3.2 Minor Variation or Type IB:
Variations which are not Type IA nor Type II or an extension. Such variations must be notified before implementation but do not require a formal approval. MAH must wait for 30 days to ensure that notification is deemed acceptable.
Major Variation or Type II: Variations which may have a significant impact on the quality, safety or efficacy of the product. Requires prior approval before implementation.
Extension: Changes to a marketing authorization listed in annex I of Commission Regulation (EC) No 1234/2008 are regarded as extensions
Extension applications include changes to active substance (by different salt/ester complex/derivative with same therapeutic moiety), strength, pharmaceutical form and route of administration
Urgent Safety Restrictions:
An interim change to product information concerning particularly one or more of the items in the summary of product characteristics, the indications, posology, contra-indication, warnings, target species and withdrawal periods due to new information having a bearing on safer use of medicinal product.
Grouping of Variations:
It is possible to group variations of different categories and submit them under a single application form to the same relevant authority. This is permissible where variations are covered under the cases listed in annex III to the variation's regulation. [4]
4. Handling of Unexpected Deviations (Unplanned Changes)
Unexpected deviations, often referred to as unplanned changes, represent a significant challenge in pharmaceutical manufacturing and regulatory compliance. These deviations occur when a product batch is manufactured or tested in a manner that does not fully conform to the registered details in the Marketing Authorization (MA) or approved Good Manufacturing Practices (GMP) documentation.
As per Eudralex Volume 4, Annex 16 – “Certification by a Qualified Person and Batch Release”, Section 3: Handling of Unexpected Deviations, a Qualified Person (QP) may still certify a batch for release provided that registered specifications for active substances, excipients, packaging materials, and the finished product are met, and the deviation does not negatively impact product quality, safety, or efficacy.
Key Requirements for Managing Unplanned Changes:
1. Thorough Investigation
The root cause of the deviation must be identified and corrective action implemented. Documentation of the investigation must be maintained as part of the batch record.
2. Quality Risk Management (QRM) Assessment
A structured QRM approach, as described in Part III of the GMP Guide, must be used to evaluate:
Whether the deviation has any impact on the product's quality, safety, or efficacy.
Whether the affected batch(es) should be included in ongoing stability programs.
For biological products, whether the deviation may affect clinical performance or immunogenicity.
3. Regulatory Notification
If the deviation is considered to represent a change to the MA, a variation application may be required for continued production under the modified conditions.
4. QP Responsibility and Batch Certification
Even if multiple QPs are involved, the QP responsible for final batch release must be aware of and assess the impact of any deviation. Certification should only proceed if it can be justified based on QRM and supporting evidence.
Example:
If a product is manufactured with a slight process parameter deviation (e.g., extended blending time), but the batch meets all final product specifications:
· The QP can consider certifying the batch after risk assessment and investigation.
· However, if the deviation becomes routine, a Type IB or Type II variation may be needed to formally update the MA to reflect the new process condition.
5. Summary of variations and anticipated implementation dates in Europe
A: Administrative changes:
o Change in the name and/or address of the MA holder – Type IAIN
o Change in the name and/or address of the Manufacturer of the finished product including quality control site – Type IAIN
o Deletion of manufacturing sites (including for an active substance, intermediate or finished product, packaging site, manufacturer responsible for batch release, site where batch control takes place, or supplier of a starting material, reagent or excipient (when mentioned in the dossier) – Type IA
B: Quality changes:
Active substance manufacture
Change in the manufacturer (including where relevant quality control sites) of the active substance, where no Ph. Eur. Certificate of Suitability is part of the approved dossier
Introduction of a new manufacturer of the active substance that is supported by an ASMF (Active substance master file) – Type II
Change in batch size (including batch size ranges) of active substance or intermediate
Up to 10-fold increase compared to the currently approved batch size – Type IA
More than 10-fold increase compared to the currently approved batch size – Type IB
5.1 Finished product description & composition
Change or addition of imprints, bossing or other markings including replacement, or addition of inks used for product marking. [5]
Change in imprints, bossing or other markings – Type IAIN
Changes in scoring/break lines intended to divide into equal doses – Type IB
Change in coating weight of oral dosage forms or change in weight of capsule shells
Solid oral pharmaceutical forms – Type IA
Gastro-resistant, modified or prolonged release pharmaceutical forms where the coating is a critical factor for the release mechanism – Type II
5.2 Manufacture
Change in the manufacturing process of the finished product
Minor change in the manufacturing process of an immediate release solid oral dosage form or oral solutions – Type IA
Change in the manufacturing process of the finished product
Substantial changes to a mfg. process that may have a significant impact on the quality, safety and efficacy of the medicinal product – Type II
Introduction or increase in the overage that is used for the active substance – Type II
Minor change in the manufacturing process of an aqueous oral suspension – Type IB
Change in the batch size (including batch size ranges) of the finished product
Up to 10-fold compared to the currently approved batch size– Type IA
Downscaling down to 10-fold– Type IA
More than 10-fold increase compared to the currently approved batch size for immediate release – Type IB
Change to in-process tests or limits applied during the manufacture of the finished product
Tightening of in-process limits – Type IA
Addition of a new tests and limits – Type IA
Deletion of an in-process test which may have a significant effect on the overall quality of the finished product – Type II
5.3 Control of Excipients
Change in the specification parameters and/or limits of an excipient
Tightening of specification limits – Type IA
Addition of a new specification parameter to the specification with its corresponding test method – Type IA
Change outside the approved specifications limits range – Type II
Deletion of a specification parameter which may have a significant effect on the overall quality of the finished product – Type II
From TSE risk material to vegetable or synthetic origin
For excipients or reagents not used in the manufacture of a biological / immunological active substance or in a biological / immunological medicinal product – Type IA
For excipients or reagents used in the manufacture of a biological / immunological active substance or in a biological / immunological medicinal product – Type IB
Change or introduction of a TSE risk material or replacement of a TSE risk material from a different TSE risk material, not covered by a TSE certificate of suitability – Type II [6]
5.4 Control of Finished product
Change in the specification parameters and/or limits of the finished product
Tightening of specification limits – Type IA
Addition of a new specification parameter to the specification with its corresponding test method – Type IA
Deletion of a non-significant specification parameter (e.g. deletion of an obsolete parameter – Type IA
Deletion of a specification parameter which may have a significant effect on the overall quality of the finished product – Type II
Change in test procedure for the finished product
Minor changes to an approved test procedure – Type IA
Deletion of a test procedure if an alternative method is already authorized – Type IA
Other changes to a test procedure (including replacement or addition) – Type IB[6]
5.5 Container closure system
Change in immediate packaging of the finished product
Qualitative and quantitative composition
Solid pharmaceutical forms – Type IA
Semi-solid and non-sterile liquid pharmaceutical forms – Type IB
Sterile medicinal products and biological / immunological medicinal products – Type II
The change relates to a less protective pack where there are associated changes in storage conditions and/or reduction in shelf life – Type II
Type of container
Solid, semi-solid and non-sterile liquid pharmaceutical forms – Type IB
Sterile medicinal products and biological/immunological medicinal products – Type II
Change in the specification parameters and/or limits of the immediate packaging of the finished product
Tightening of specification limits – Type IA
Addition of a new specification parameter to the specification with its corresponding test method – Type IA
Change in shape or dimensions of the container or closure (immediate packaging)
Non-sterile medicinal products – Type IA
Sterile medicinal products – Type IB
Change in pack size of the finished product
Change in the number of units (e.g. tablets, ampoules, etc.) in a pack
Change within the range of the currently approved pack sizes – Type IAIN
Change outside the range of the currently approved pack sizes – Type IB
Deletion of a pack size(s) – Type IA
Change in the fill weight/fill volume of sterile multi-dose (or single-dose, partial use) parenteral medicinal products, and biological/immunological multi-dose parenteral medicinal products– Type II
Change in the fill weight/fill volume of non-parenteral multi-dose (or single-dose, partial use) products– Type IB
Change in supplier of packaging components or devices (when mentioned in the dossier)
Deletion of a supplier – Type IA
Replacement or addition of a supplier – Type IA [7]
5.6 Stability
Change in the shelf-life or storage conditions of the finished product
Reduction of the shelf life of the finished product
As packaged for sale – Type IAIN
After first opening – Type IAIN
After dilution or reconstitution – Type IAIN
Extension of the shelf life of the finished product
As packaged for sale (supported by real time data) – Type IB
After first opening (supported by real time data) – Type IB
After dilution or reconstitution (supported by real time data) – Type IB
Extension of the shelf-life based on extrapolation of stability data not in accordance with ICH guidelines– Type II
Change in storage conditions of the finished product or the diluted / reconstituted product – Type IB [8]
CONCLUSIONS
To prevent the potential that modifications to a pharmaceutical product may give birth to public health issues, the European variations processes were developed. It should be remembered, nonetheless, that any modification to the documentation, any deletion, and/or any alteration to the content will result in a variation procedure. In some circumstances, it is beneficial to evaluate data that is important to the agency and update obsolete or too extensive paperwork to prevent additional deviations. Analytical validation protocols included in the documentation, for example, may be important for inspections but will result in a combination of variations in the regulatory framework for an approved drug product.
References
01. Article 5 procedure: Regulatory and procedural guidance | European Medicines Agency. (n.d.). Retrieved February 7, 2023, from https://www.ema.europa.eu/en/human-regulatory/post-authorisation/variations/article-5-procedure-regulatory-procedural-guidance
02. Type-IA variations: questions and answers | European Medicines Agency. (n.d.). Retrieved February 7, 2023, from https://www.ema.europa.eu/en/veterinary-regulatory/post-authorisation/variations/variations-guidance-under-regulation-ec-no-1234-2008-regulation-eu-no-712-2012/type-ia-variations-questions-answers
03. EU Variation Guidance New | PDF | European Commission | Member State Of The European Union. (n.d.). Retrieved February 7, 2023, from https://www.scribd.com/document/441463855/EU-Variation-Guidance-New#
04. MINISTRY OF HEALTH - Welcome to our Website. (n.d.). Retrieved February 7, 2023, from https://www.moh.gov.cy/moh/moh.nsf/index_gr/index_gr?OpenDocument
05. Державний експертний центр Міністерства охорони здоров’я України. (n.d.). Retrieved February 7, 2023, from https://www.dec.gov.ua/
06. Brezovska, M., Acevska, J., Nakov, N., Ugrinova, L., & Tonic Ribarska, J. (2022). Comparative analysis of the regulatory framework for postmarketing authorization changes in EU and USA. Macedonian Pharmaceutical Bulletin, 68(03), 205–206. https://doi.org/10.33320/MACED.PHARM.BULL.2022.68.03.098
07. Khanna, B. (2018). Pharmaceutical Regulations in European Union. Pharmaceutical Medicine and Translational Clinical Research, 175–213. https://doi.org/10.1016/B978-0-12-802103-3.00012-2
08. Zuckerman, D. M., Brown, P., & Nissen, S. E. (2011). Medical Device Recalls and the FDA Approval Process. Archives of Internal Medicine, 171(11), 1006–1011. https://doi.org/10.1001/archinternmed.2011.30
Acknowledgements
I would like to express my special gratitude and thanks to my organization (JSS College of Pharmacy Mysore) for giving me the opportunity of designing and drafting the manuscript of this review article.
Conflict of Interest
All authors declare that they have no conflicts of interest.
Authors
Arunodaya B S,1 Gaganashree T V*, Balamuralidhara V2, Chinmayee U Gowda
1Research scholar, Department of Pharmaceutics, Pharmaceutical Regulatory Affairs Group, JSS College of Pharmacy, JSS Academy of Higher Education Research, Mysuru, Karnataka.
2 Head of the Department of Pharmaceutics, JSS College of Pharmacy, JSS Academy of Higher Education Research, Mysuru, Karnataka
Corresponding Author: Gaganashree T V
Email: Gaganashreeraju@gmail.com



Comments